| Меню сайта |
 |
|
|
 |
| Мини-чат |
|
|
|
|
| Статистика |
|
| |
| |
|
20-летний солдат с 3-дневной темной жгучей сыпью
| |
| wahib |
Дата: Суббота, 07.03.2009, 18:36 | Сообщение # 1 |
 Египет Группа: Пользователи
Сообщений: 30
Статус: Offline |
A 20-Year-Old Soldier With a 3-Day History of a Dark, Burning Rash Background A 20-year-old active-duty male soldier presents to the emergency department (ED) of a military hospital complaining of a 3-day history of a dark-red "burning rash." The rash started at his sock line and, over the course of the past 2 days, has spread proximally up his thighs. It is not present on his abdomen or back, but it has spread to his hands over the past day. The patient also developed a sore throat and a scratchy voice the day before presentation, without odynophagia. He was in the North Carolina woods as part of his infantry training, which is supplemental training after boot camp. He reports having spent 20+ hours per day for the past several days in a foxhole (a hole in the ground that soldiers use for protection). The patient reports fatigue only related to his level of activity and lack of sleep during training. He is not taking any medications, he has no allergies, and he has smoked 5-10 cigarettes daily for the past year. He also reports drinking approximately 5-10 beers with his comrades 1-2 times per month before starting his training. The patient notes some right ankle swelling and pain that started after a 12.43 mi (20 km) field hike with a 60 lb (27.22 kg) rucksack on his back. The pain and swelling began 1 month before admission and has worsened in the past few days. He has had no pruritus, fevers, chills, abdominal pain, diarrhea, changes in urine color, or dysuria, as well as no new sexual contacts and no recent animal or insect bites. No one else in his military unit has reported similar skin findings. The patient had a minor motorcycle accident 18 months ago, with a right leg laceration that required suture repair. He has no significant family history.
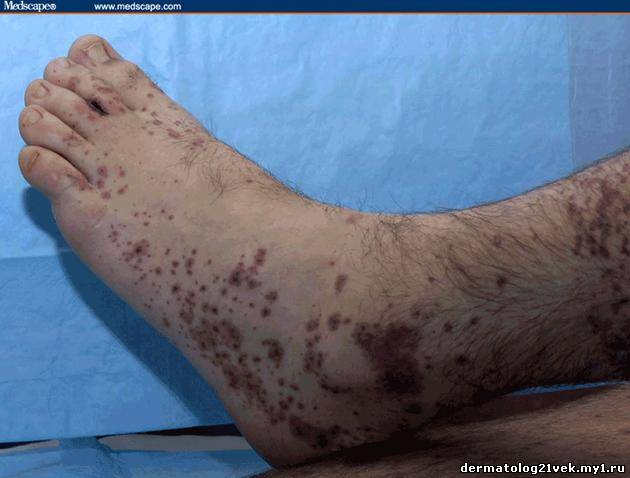
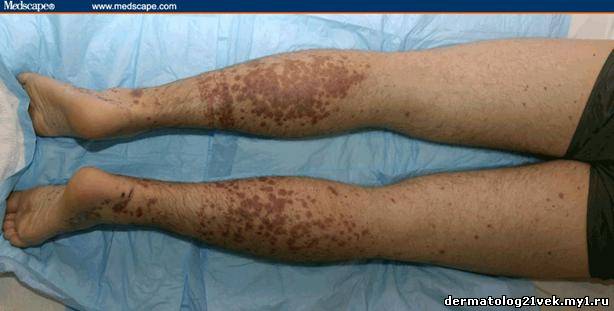
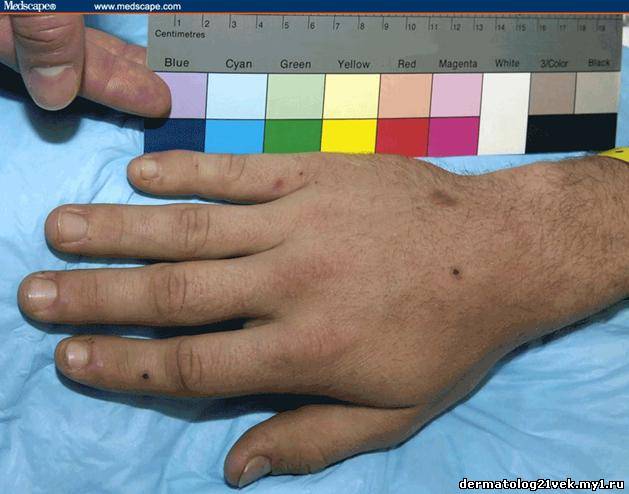
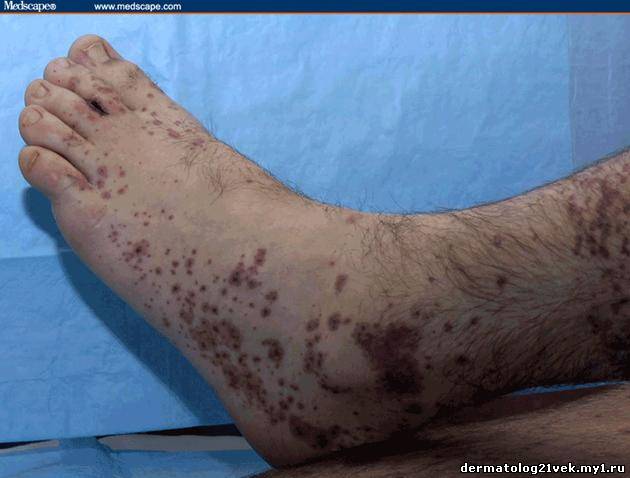
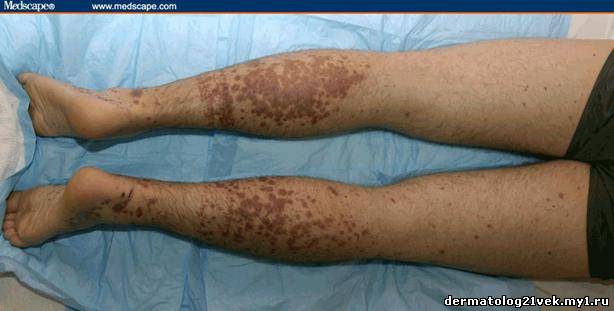
On physical examination, his vital signs demonstrate a strong pulse, with a regular rhythm and a rate of 58 bpm, blood pressure of 123/60 mm Hg, weight of 165.3 lb (75 kg), and oral temperature of 98.1°F (36.7°C). He is a very fit, well-developed white male in no apparent distress. The pharynx shows no erythema or exudate, and the neck examination demonstrates no tenderness to palpation or lymphadenopathy. The cardiac examination is notable for a point of maximum intensity at the 5th left interspace, but no murmurs, gallops, or rubs are appreciated. The patient's pulses are strong bilaterally. The respiratory examination reveals lungs clear to auscultation bilaterally. The abdominal examination is unremarkable, and the stool test is negative for occult blood. The skin examination show lesions ranging in diameter from 2 mm to 10 cm (see Figures 1-3). The macules and plaques are nonblanchable, and most of them are concentrated in the posterior calves, the palms, and the soles of the feet. No macules or plaques are noted on the face, chest, or back. The lesions are not tender to palpation or warm to the touch. His hands and right ankle exhibit 1+ nonpitting edema. The patient has a scar on his right leg from his motorcycle accident that is well-healed, with no fluctuance or erythema. The neurologic examination is unremarkable.
A urinalysis shows 2+ protein and 2+ blood, with 25-50 red blood cells (RBCs) per high-power field on microscopy. His coagulation studies at admission include a prothrombin time (PT) of 12.3 s, an international normalized ration (INR) of 1.08, and a partial thromboplastin time (PTT) of 25.5 s. The patient's blood urea nitrogen (BUN) is 24 mg/dL (8.59 mmol/L) and his creatinine value is 1.1 mg/dL (97.2 µmol/L). The complete blood count (CBC) shows a white blood cell (WBC) count of 7.4 × 103/µL (7.4 × 109/L), a hemoglobin (HGB) of 12.4 g/dL (124 g/L), a hematocrit (HCT) of 37.1% (0.371), and a platelet count of 195 × 103/µL (195 × 109/L), with a normal smear. Skin punch biopsies of 2 of the lesions are obtained that demonstrate small-vessel leukocytoclastic vasculitis. Immunofluorescence of the skin biopsies demonstrates a weak linear pattern at the dermal-epidermal junction for immunoglobulin G (IgG), immunoglobulin A (IgA), and complement 3 (C3). 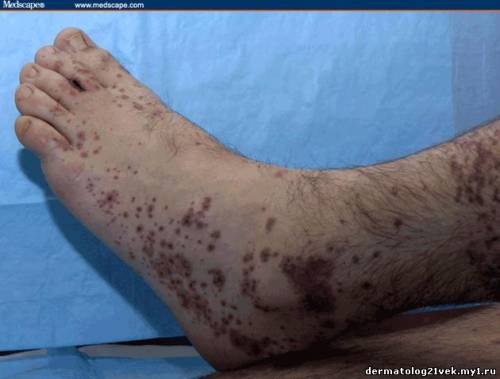 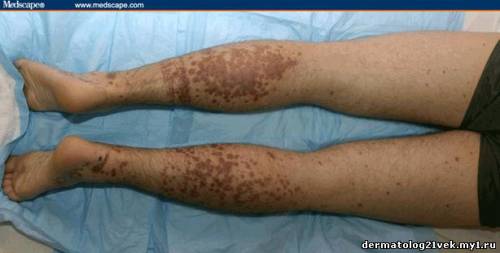 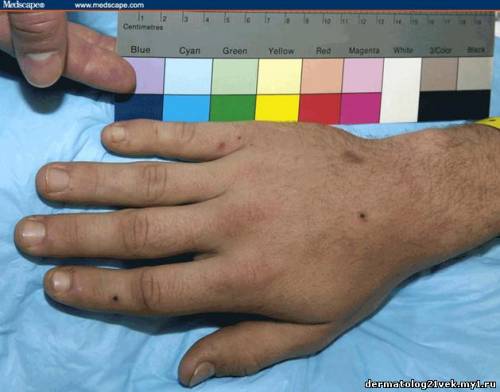 What is the diagnosis? Hint: The patient is presenting with nonblanchable lesions after reporting an antecedent sore throat.
Henoch-Schönlein purpura (HSP)
Mixed cryoglobulinemia
Polyarteritis nodosum
Systemic lupus erythematosus
Hemolytic uremic syndrome (HUS) Your Colleagues Responded:
Henoch-Schönlein purpura (HSP) Correct Answer 62%
Mixed cryoglobulinemia 6%
Polyarteritis nodosum 7%
Systemic lupus erythematosus 6%
Hemolytic uremic syndrome (HUS) 17% Discussion    Figure 1. Figure 2. Figure 3. There exist several guidelines for the diagnosis of Henoch-Schönlein purpura.[1] The American College of Rheumatology (ACR) requires 4 criteria (published in 1990) for diagnosing Henoch-Schönlein purpura[2]: palpable purpura, patient age of 20 years or less at onset, bowel angina, and the presence of granulocytes in the vessel walls. More recently, in 2006, the European League Against Rheumatism (EULAR) and the Pediatric Rheumatology Society published their own Henoch-Schönlein purpura criteria. These include palpable purpura as a mandatory criterion, with at least one of the following conditions: diffuse abdominal pain, predominant IgA deposition (confirmed on biopsy), acute arthritis in any joint, and renal involvement (as evidenced by hematuria and/or proteinuria).[3] According to the older ACR criteria, the diagnosis of Henoch-Schönlein purpura for our patient could be called into question, as he exhibited no bowel angina; however, according to the more recent EULAR criteria, our patient fits the diagnosis of Henoch-Schönlein purpura quite well, as he has palpable purpura, predominant IgA deposition confirmed on biopsy, a monoarticular arthralgia in his right ankle, and renal involvement.
The sine qua non for the diagnosis of Henoch-Schönlein purpura is a skin biopsy finding of IgA deposition at the dermal-epidermal junction. A skin biopsy with immunofluorescence studies is recommended if clinical suspicion of Henoch-Schönlein purpura exists.[4] The etiology of the disease remains unknown, but it is understood to be an autoimmune response (usually to an upper respiratory infection).
Other items in the differential diagnosis include many of the etiologies of palpable purpura. An algorithmic attempt has been made (see Table 1) to delineate these etiologies, but classification remains difficult.[5] Palpable purpura is a classic skin manifestation of cutaneous vasculitis, but there are many other entities that can cause it.[6] Additional information can be gleaned from the pathologic diagnosis of small vessel vasculitis, which also carries its own differential (see Table 2).[7] The intersection of these 2 differentials consists of the following set of diagnoses: mixed cryoglobulinemia, vasculitis associated with collagen vascular disease (ie, systemic lupus erythematosus, rheumatoid arthritis), and Henoch-Schönlein purpura.
Mixed cryoglobulinemia was easily ruled out, as the patient's serum cryoglobulin test and complement levels were a C3 of 118 mg/dL (1.18 g/L; normal range, 79-152 mg/dL), a C4 of 26 mg/dL (0.26 g/L; normal range, 16-38 mg/dL), complement CH50 of 53.0 U/mL (53 kU/L; normal range 22-60 U/mL), and a negative serum cryoglobulin examination. Another diagnosis that was readily excluded was systemic lupus erythematosus. Our patient had only 1 arthralgia, but no fevers, myalgia, or malaise were noted; only 1/11 American Rheumatologic Association criteria for the diagnosis of lupus were met.[8] To satisfy the clinical diagnosis of systemic lupus erythematosus, 4/11 American Rheumatologic Association criteria are required.[8] Although the patient meets the histopathologic criteria for solitary IgA nephropathy, as the renal biopsy showed diffuse proliferative endocapillary glomerulonephritis with increased mesangial matrix and cellularity, capillary wall thickening, and lobular accentuation on light and electron microscopy with strong (3+) mesangial staining for IgA and fibrinogen, as well as lappa and lambda light chains (2-3+) present on immunofluorescence microscopy, he also exhibited some of the cardinal clinical features of Henoch-Schönlein purpura at presentation. He denied having had blood in his urine on prior urinalyses or a history of gross blood urine, lessening the possibility of IgA nephropathy. After further inquiry, it was confirmed that the patient had no family history of renal disease and, as such, it is unlikely that he has familial IgA nephropathy. Polyarteritis nodosum (PAN) could be considered in the differential diagnosis, but this patient displayed none of the classic signs or symptoms of fever, abdominal pain, nausea, and weight loss. Also common to PAN is the is involvement of small- and medium-sized arteries, as well as subcutaneous nodules (not palpable purpura). Joint pain, however, is very common in PAN. PAN may be associated with hepatitis B or C, but this patient's hepatitis panel was negative. A hemorrhagic diathesis was quickly ruled out with the normal coagulation studies. Lyme disease, in contrast to Henoch-Schönlein purpura, more commonly presents with the typical target lesions of erythema migrans and a positive Lyme titer (and not with a purpuric rash). This patient had a negative Lyme antibody examination, although this finding alone does not exclude the possibility of the disease. Multiple arthralgias are more common in Lyme disease, and they seldom present with abdominal pain. Hemolytic uremic syndrome (HUS) should be considered in the differential diagnosis, but it can be easily recognized by the presence of microangiopathic anemia and is distinguished from Henoch-Schönlein purpura by the absence of purpura and arthralgias.
Henoch-Schönlein purpura is an autoimmune, self-limited IgA-mediated vasculitis that most commonly presents in children 5 years of age. In fact, 95% of cases present in children <10 years of age.[9] There is a 2:1 distribution between males and females.[10] The overall incidence is 0.14 cases per 1000 population in children aged 2-14.[10] This patient is in a relatively uncommon demographic for the disease. The most common signs and symptoms of the disease are palpable purpura (100%), abdominal pain (63%), gastrointestinal bleeding (33%), arthralgia (82%), nephritis (40%), and hematuria.[11] Risk factors associated with a poor prognosis for renal survival include nephrotic syndrome, decreased factor XIII activity, hypertension, and renal failure at onset.[9] It is known that severe Henoch-Schönlein purpura–associated nephritis and progression to nephrotic syndrome are associated with a poorer prognosis.[9]
The mainstays of treatment for Henoch-Schönlein purpura are observation, intravenous hydration, and steroid therapy. Most cases are self-limited and resolve within 4 weeks.[12,13] Cyclophosphamide is usually reserved for severe, systemic forms of vasculitis, and appropriate consultation with a nephrologist is recommended.[16] Chronic kidney disease and end-stage renal disease are known complications of Henoch-Schönlein purpura, with incidence rates of 5% and 1.5%, respectively.[12] It should be noted that corticosteroids should only be instituted after consultation with a nephrologist, as they are of questionable benefit.[13,14] In fact, only 1 randomized controlled clinical trial has been performed evaluating the use of corticosteroids in treating Henoch-Schönlein purpura as renal protective agents. Huber et al found that early prednisone therapy initiation did not ward off chronic kidney disease at 1 year[14]; however, there has been a case report of successful treatment of an adult patient with crescentic glomerulonephritis (from Henoch-Schönlein purpura) with 9 rounds of double-filtration plasmapheresis and concomitant plasma replacement.[15]
Our patient was admitted to the hospital ward and vigorously volume-expanded with normal saline at 250 cc/h. His creatinine clearance was 182.7 mL/min (3.05 mL/s; this is within normal limits). Also notable was his antistreptolysin O (ASO) titer of 722 Todd U/mL (normal range, 0-117 Todd U/mL). Even with his severe skin manifestations, the severity of our patient's Henoch-Schönlein purpura nephritis on admission was low. The patient's positive ASO titer pointed to a recent presumed streptococcal pharyngitis. The skin biopsy report was significant for small-vessel leukocytoclastic vasculitis. The immunofluorescence study of the sample demonstrated a weak linear pattern at the dermal-epidermal junction, with IgG, IgA, and C3, which was consistent with the diagnosis of Henoch-Schönlein purpura. On admission, the patient's protein-to-creatinine ratio was 5.5, consistent with nephrotic-range proteinuria, or >3.5 grams of protein per day. A normal protein-to-creatinine ratio is <0.2.[17] Although the nephrology service was consulted early in the clinical course and the patient was started on high dose intravenous methylprednisolone therapy at 1 gram divided twice a day on hospital day 1, his condition rapidly worsened. By hospital day 4, the patient's serum creatinine was 1.1 mg/dL (97.2 µmol/L) and his protein-to-creatinine ratio had increased to 15.1. His urine volume was 1850 mL on that day, with 24 h protein and creatinine excretion totaling 35.8 grams and 2384 mg, respectively. The patient, while still normotensive, had developed prominent edema consistent with nephrotic syndrome. As his renal status had worsened significantly, he was transferred to a tertiary care center for further nephrology care. A subsequent renal biopsy demonstrated diffuse proliferative endocapillary glomerulonephritis consistent with IgA nephropathy, or Henoch-Schönlein purpura. Mild chronic tubulointerstitial disease and mild hyaline arteriolar sclerosis were noted as well. A regimen of cyclophosphamide was initiated, and the patient deteriorated to end-stage renal disease by the time this case was compiled. Table 1: Etiology of Purpura • Intravascular (usually nonpalpable) Alterations in platelet formation, destruction, or function
• Drugs (aspirin, methyldopa)
• Idiopathic thrombocytopenic purpura
• Thrombotic thrombocytopenic purpura
• Disseminated intravascular coagulation
• Infection (especially Neisseria, Rickettsia, Staphylococcus)
• Splenic sequestration
• Radiation therapy
• Myelofibrosis
• Myeloproliferative disorders
Vascular (usually palpable) Inflammatory vascular causes
• Cutaneous vasculitis
• Purely cutaneous vasculitis (eg, secondary to medications)
• Henoch-Schönlein purpura
• Polyarteritis nodosa
• Granulomatous vasculitis (Wegener granulomatosis, Churg-Strauss vasculitis)
• Cutaneous vasculitis associated with a collagen
• Vascular disease (eg, systemic lupus erythematosus, rheumatoid arthritis)
• Giant cell arteritis
• Mixed cryoglobulinemia
• Hyperglobulinemic purpura
Noninflammatory vascular causes
• Trauma
• Subacute bacterial endocarditis
• Other embolic diseases
• Amyloidosis
• Extravascular and miscellaneous (may be palpable or nonpalpable) Corticosteroids
• Toxins and venoms
• Senile purpura
• Scurvy
• Valsalva maneuver
• Pseudopurpura (Sweet syndrome, cherry angiomas, angiokeratoma, Kaposi sarcoma)
Derived from: Schreiner DT. Purpura. Dermatol Clin. 1989;7(3):481-90 Table 2: Clinical Signs of Necrotizing Vasculitis with Respect to Vessel Size Involved Small Vessels
Signs Diseases
• Urticaria Hypersensitivity vasculitis
• Palpable Purpura Henoch-Schönlein purpura
• Essential Mixed Cryoglobulinemia
• Vasculitis associated with connective tissue disease
• Vasculitis associated with malignancy
• Serum sickness and serum sickness-like reactions
• Nodules, bullae, or ulcers Chronic urticaria (urticarial vasculitis)
• Urticarial prodrome of acute hepatitis type B infection
Large Vessels
Signs Diseases
• Subcutaneous nodules, ulceration, and, ecchymoses Polyarteritis nodosa
• Churg-Strauss syndrome
• Wegener granulomatosis
• Giant cell (temporal) arteritis
Derived from: Habif, TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 4th ed. St. Louis: Mosby; 2004. Test If the clinician is clinically suspicious of Henoch-Schönlein purpura in a patient, which test is most likely to give a definitive diagnosis?
Urine protein electrophoresis
Skin biopsy
Serum complement levels
24 h urine protein
CBC Which of the following statements about Henoch-Schönlein purpura is true?
Henoch-Schönlein purpura treatment requires corticosteroids.
Henoch-Schönlein purpura treatment requires dialysis.
Henoch-Schönlein purpura treatment requires plasmapheresis.
Most cases of Henoch-Schönlein purpura spontaneously resolve within 1 month.
Most cases of Henoch-Schönlein purpura treatment are steadily progressive and typically manifest significant renal pathology. If the clinician is clinically suspicious of Henoch-Schönlein purpura in a patient, which test is most likely to give a definitive diagnosis?
Your Colleagues Responded:
Urine protein electrophoresis 5%
Skin biopsy Correct Answer 81%
Serum complement levels 7%
24 h urine protein 3%
CBC 2%
Which of the following statements about Henoch-Schönlein purpura is true?
Your Colleagues Responded:
Henoch-Schönlein purpura treatment requires corticosteroids. 17%
Henoch-Schönlein purpura treatment requires dialysis. 0%
Henoch-Schönlein purpura treatment requires plasmapheresis. 2%
Most cases of Henoch-Schönlein purpura spontaneously resolve within 1 month. Correct Answer 70%
Most cases of Henoch-Schönlein purpura treatment are steadily progressive and typically manifest significant renal pathology. 7%
Сообщение отредактировал vachib - Суббота, 07.03.2009, 18:50 |
| |
| |
| Наталья |
Дата: Суббота, 07.03.2009, 18:56 | Сообщение # 2 |

Группа: Администраторы
Сообщений: 229
Статус: Offline |
20-летний солдат с 3-дневной темной жгучей сыпью
Общее состояние 20-летний солдат срочной службы предъявляет жалобы чрезвычайной комиссии военной больницы на 3-дневный анамнез темно-красной жгучей сыпи. Сыпь, возникшая первоначально в области стоп, в течение 2 дней распространилась проксимально на область бедер. Она отсутствует в области живота и спины, но за прошедший день распространилось на область рук. У пациента также развилось воспаление горла, осиплость голоса за день до появления высыпаний, без болезненности при глотании. Он был на учениях в лесах Северной Каролины, которое является дополнительным обучением после учебного лагеря. Он сообщает, что проводил более 20 часов в день в течение прошлых нескольких дней в окопах (отверстие в земле, которое солдаты используют для защиты). Пациент сообщает о переутомлении, связанном с физической нагрузкой и недостатком сна во время обучения. Он не принимал никаких препаратов, не имеет аллергии в анамнезе и курил 5-10 сигарет ежедневно в течение прошлого года. Он также сообщает о питье приблизительно 5-10 кружек пива с товарищами 1-2 раза в месяц перед началом обучения. Пациент обращает внимание на отечность и боль в области правой лодыжки, которая началась после 20-километрового полевого марш-броска с 27.22-килограммовым рюкзаком на спине. Боль и отёчность начались за 1 месяц до госпитализации и ухудшились за последние несколько дней. У него не было зуда, лихорадки, озноба, боли в брюшной полости, диареи, изменения цвета мочи или дизурии, а также никаких новых сексуальных контактов и контакта с животными или укусов насекомых. Никто еще в его военной части не сообщал о подобных высыпаниях. Пациент имел незначительный несчастный случай на мотоцикле 18 месяцев назад с травмой правой ноги, которая требовала ушивания раны. Он не имеет никаких особенностей семейного анамнеза.
При физическом осмотре: сильный пульс, с правильным ритмом и темпом 58 раз в мин., кровяное давление 123/60 мм рт ст, вес 75 кг, и оральная температура 36.7°C. Он - здоровый, хорошо развитый белый мужчина без признаков истощения. Зев не гиперемирован, без отделяемого, шея безболезненна при пальпации, лимфатические узлы не увеличены. Сердечная экспертиза в точке максимальной интенсивности в 5-ом левом межреберье не выявила шумов, нарушения ритма. Пульс сильный с двух сторон. Дыхательная экспертиза: в легких дыхание ясное с двух сторон. Брюшная экспертиза без особенностей, исследование анализа кала на скрытую кровь отрицательно. При осмотре кожи - рассеянная сыпь диаметром от 2 мм до 10 см (см. Фото 1-3). Пятна и бляшки – не бледнеющие при надавливании, максимально сконцентрированы на задней поверхности голеней, ладонях и подошвах. Пятна и бляшки отсутствуют на лице, груди или спине. Повреждения безболененны припальпации и не горячие на ощупь. Его руки и правая лодыжка отечны, при пальпации не остается ямка. Пациент имеет заживший шрам на правой ноге от несчастного случая на мотоцикле, без флюктуации или эритемы. Неврологическая экспертиза без особенностей.
Анализ мочи: белок2 + , эритроциты 25-50 в поле зрения. Исследование коагуляции при госпитализации: протромбиновое время 12.3 с, частичное тромбопластиновое время
25.5 с. Мочевина крови 8.59 mmol/), креатинин - 1.1 mg/dL . Общий анализ крови : лейкоциты 7.4 Ч 109/L, гемоглобин 124 g/L, гематокрит 0.371, количество тромбоцитов 195 Ч 109/L. Пункционная биопсия кожи из 2 кожных элементов, демонстрирует лейкоцитокластический васкулит. Иммунофлюоресценция биопсия кожи демонстрирует снижение в дермо-эпидермальном соединении иммуноглобулина G (IgG), иммуноглобулина А (IgA), и комплемента 3 (C3). 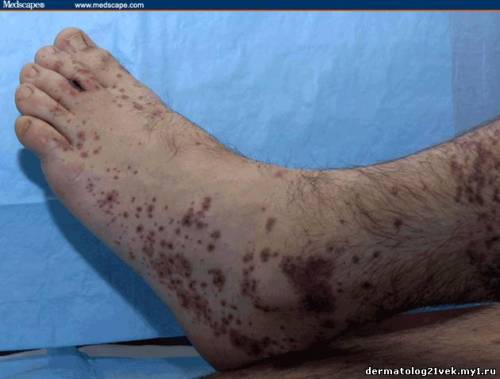 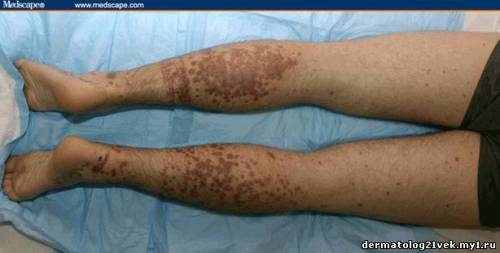 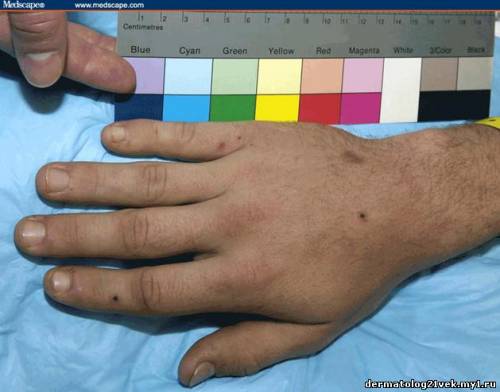 Каков диагноз? Намек: пациент жалуется на небледнеющие высыпания после в горле.
пурпура Шейнляйн - Геноха (HSP)
Смешанная криоглобулинемия
Узловой периартериит
Системная красная волчанка
Гемолитический уремический синдром (HUS) Ваши Коллеги Отвечали:
пурпура Шейнляйн - Геноха (HSP) Правильный Ответ 62 %
Смешанная криоглобулинемия 6 %
Узловой периартериит 7 %
Системная красная волчанка 6 %
Гемолитический уремический синдром 17 % Обсуждение    Фото 1. Фото 2. Фото 3. Существует несколько руководящих принципов для постановки диагноза пурпуры Шейнляйн - Геноха. Американский Колледж Ревматологии (ACR) требует 4 критерия (изданные в 1990) для того, чтобы диагностировать пурпуру Шейнляйн - Геноха: пальпируемая пурпура, начало заболевания в 20 летнем возрасте или ранее, колит и гранулоцитарный инфильтрат в стенках сосудов. Позже, в 2006, Европейская Лига по Борьбе Против Ревматизма (EULAR) и Педиатрическое Общества Ревматологии издала собственные критерии пурпуры Шейнляйн - Геноха. Они включают пальпируемую пурпуру, как обязательный критерий, в сочетании с по крайней мере одним из следующих состояний: распространенная брюшная боль, преобладающее смещение IgA (подтвержденное на биопсии), острый артрит в любом суставе и почечные нарушения (гематурия и/или протеинурия). Согласно старым ACR критериям, диагноз пурпуры Шейнляйн - Геноха для нашего пациента мог быть подвергнут сомнению, поскольку у него не было никаких жалоб со стороны кишечника; однако, согласно более недавним EULAR критериям, наш пациент подпадает под диагноз пурпуры Шейнляйн - Геноха весьма хорошо, поскольку он имеет пальпируемую пурпуру, преобладающее смещение IgA, подтвержденное на биопсии, моносуставнуя артралгию в области правой лодыжки и почечные симптомы.
Непременное условие для постановки диагноза пурпуры Шейнляйн - Геноха – обнаружение при биопсии кожи смещения IgA в дермо-эпидермальном соединении. Биопсия кожи с иммунофлюоресцентным исследованием рекомендуется, если существует клиническое подозрение на пурпуру Шейнляйн - Геноха. Этиология болезни остается неизвестной, но как предполагается, является аутоиммунным ответом (обычно к инфекции верхних дыхательных путей).
Другие пункты в диф. диагностике включают многие причины пальпируемой пурпуры. Была сделана попытка очертить алгоритм (см. Таблицу 1) этих причин, но классифицировать остается трудным. Пальпируемая пурпура - классическое проявление кожного васкулита, но есть много других состояний, которые могут ее вызвать. Дополнительная информация может быть получена из патологического диагноза поверхностных васкулитов, который также должен дифференцироватся. (см. Таблицу 2). Пересечение этих 2 дифференциалов состоит из следующего набора диагнозов: смешанная криоглобулинемия, васкулиты при системных заболеваниях соединительной ткани (то есть, системная красная волчанка, ревматоидный артрит), и пурпура Шейнляйн - Геноха.
Смешанная криоглобулинемия была легко исключена, поскольку исследование криоглобулинов в сыворотке крови пациента отрицательно, и уровень комплемента C3 -118 mg/dL (нормальный диапазон, 79-152 mg/dL), C4- 26 mg/dL ( нормальный диапазон, 16-38 mg/dL), комплемент CH50 53.0 U/mL ( нормальный диапазон 22-60 U/mL).
Другой диагноз, который был с готовностью исключен, был системная красная волчанка. Наш пациент имел артралгию только одного сустава, но повышение температуры, миалгия или общее недомогание не были отмечены; только 1 из11 критериев для диагноза Красная волчанка Американской Ассоциации Ревматологии lupus были встречены. Чтобы поставить клинический диагноз Системной красной волчанки, требуются 4 из 11 критериев Американской Ассоциации Ревматологии. Хотя у пациента встречаются гистопатологические критерии изолированной IgA- нефропaтии, поскольку почечная биопсия показала диффузный пролиферативный эндокапиллярный гломерулонефрит, с утолщением капиллярных стенок и выделением долек при световой и электронной микроскопии с сильным (3 +) окрашиванием мезенхимы для IgA и фибриногена, а также показала некоторые из кардинальных клинических особенностей пурпуры Шейнляйн – Геноха. Он отрицал наличие клеток крови в моче в предшествующем анализе мочи или грубой примеси крови в моче, что уменьшает возможность IgA нефропaтии. После дальнейшего изучения было подтверждено, что пациент не имел в семейном анамнезе почечных болезней и, также, маловероятно, что он имеет семейную IgA нефропaтию. Узловой периартериит можно было бы рассмотреть в диф. диагнозе, но этот пациент не имел ни одного из классических признаков: лихорадки, брюшной боли, тошноты и потери веса. Кроме того, обычно узловой периартериит вовлекает мелкие и средние артерии, и образует подкожные узлы (не пальпируемая пурпура). Суставные боли, однако, очень характерны для узлового периартериита. Узловой периартериит может быть связан с гепатитом B или C, но анализы на гепатиты у этого пациента были отрицательны. Геморрагический диатез был быстро исключен в связи с нормальными показателями коагуляции. Болезнь Лайма, в отличие от пурпуры Шейнляйн – Геноха, обычно представлена типичными мишеневидными высыпаниями в виде мигрирующей эритемы и положительного титра антител (а не геморрагической сыпью). Этот пациент имел отрицательные анализы на наличие титра антител к болезни Лайма, хотя однократное обследование не исключает возможности этой болезни. Множественные артралгии типичны для болезни Лайма и она редко вызывает боль в животе. Гемолитический уремический синдром нужно учесть в дифференциальном диагнозе, но он может быть легко признан присутствием микроангиопатией, анемии и отличается от пурпуры Шейнляйн – Геноха отсутствием пурпуры и артралгий.
Пурпура Шейнляйн – Геноха - это аутоиммунный IgA-обусловленный васкулит, который наиболее часто возникает у детей 5- летнего возраста. Фактически, 95 % случаев возникают у детей младше 10 летнего возраста. Соотношение между мужчинами и женщинами 2:1. Заболеваемость - 0.14 случаев на 1000 детского населения в возрасте 2-14 лет. Этот пациент находится в относительно нехарактерном возрасте для болезни. Самые частые признаки болезни - пальпируемая пурпура (100%), боль в животе (63 %), желудочно-кишечные кровотечения (33 %), артралгии (82 %), нефриты (40  и гематурия. Факторы риска, связанные с плохим прогнозом для почечного выживания включают нефротический синдром, уменьшение деятельности фактора XIII, гипертония и почечная недостаточность. Известно, что тяжелый нефрит, связанный с пурпурой Шейнляйн – Геноха и прогрессирование нефротического синдрома связаны с более плохим прогнозом. и гематурия. Факторы риска, связанные с плохим прогнозом для почечного выживания включают нефротический синдром, уменьшение деятельности фактора XIII, гипертония и почечная недостаточность. Известно, что тяжелый нефрит, связанный с пурпурой Шейнляйн – Геноха и прогрессирование нефротического синдрома связаны с более плохим прогнозом.
Основа лечения пурпуры Шейнляйн – Геноха - наблюдение, внутривенная гидратация и стероидная терапия. Большинство случаев проходит самостоятельно и разрешается в течение 4 недель. Циклофосфамиды обычно используются для серьезных, системных форм васкулитов и рекомендуется соответствующая консультация нефролога. Хроническая почечная недостаточность и конечная стадия почечной болезни - известные осложнения пурпуры Шейнляйн – Геноха, с частотой 5 % и 1.5 %, соответственно. Необходимо отметить, что кортикостероиды должны быть назначены только после консультации нефролога, поскольку они имеют сомнительную пользу. Фактически, только 1 рандомизированное управляемое клиническое исследование было выполнено для оценки использования кортикостероидов в лечении пурпуры Шейнляйн – Геноха, как почечных защитных агентов. Хабер и др. установили, что раннее начало терапии преднизолоном не предотвратило развития хронической почечной недостаточности у годовалого пациента; однако, был сообщен случай успешного лечения взрослого пациента с полулунным гломерулонефритом (на фоне пурпуры Шейнляйн – Геноха) с 9 раундами двойного- фильтрационного плазмафереза и сопровозждающегося замещением плазмы крови.
Нашему пациенту до госпитализации энергично вливали физ. раствор 250 мл/час. Показатели креатинина - 182.7 мл/мин (в пределах нормы). Также был известен его титр антистрептолизина O (ASO) 722 Ед/мл (нормальный диапазон, 0-117 Ед/мл). Даже с его серьезными кожными проявлениями, вероятность развития серьезного нефрита на фоне пурпуры у нашего пациента при госпитализации была низка. Положительный ASO титр пациента указал на недавний предполагаемый стрептококковый фарингит. Гистологические признаки были характерны для лейкоцитокластического васкулита мелких сосудов. Иммунофлюоресцентное исследование образца демонстрировало слабый линейный образец в кожном-эпидермальном соединении с IgG, IgA, и C3, который был совместим с диагнозом пурпуры Шейнляйн – Геноха. При поступлении, отношение белка к креатинину пациента было 5.5, совместимо с протеинурией нефротического диапазона, или> 3.5 граммов белка в день. Нормальное отношение белка к креатинину - <0.2. Хотя пациент был проконсультирован нефрологом на ранней клинической стадии, была начата внутривенная терапия высокими дозами метилпреднизолона (1 грамм, разделенный на два приема в день), его состояние быстро ухудшалось. К 4 дню госпитализации креатинин сыворотки пациента был 1.1 mg/dL (97.2 µmol/L), и его отношение белка к креатинину увеличилось до 15.1. Его объем мочи были 1850 мл в этот день, с круглосуточным выделением белков и креатинина 35.8 граммов и 2384 мг соответственно. У пациента при нормальном давлении развился значительный отёк, совместимый с нефротическим синдромом. Поскольку его почечный статус значительно ухудшился, он был переведен в третичный центр нефрологии. Последующая почечная биопсия демонстрировала диффузный пролиферативный эндокапилярный гломерулонефрит совместимый с IgA нефропaтиейили пурпурой Шейнляйн – Геноха. Также были отмечены умеренная хроническая тубулоинтерстициальная болезнь и умеренный гиалиновый артериолярный склероз. Был начат курс циклофосфамида. Таблица 1: Этиология пурпуры • Внутрисосудистая (обычно не пальпируется)
Изменения в формировании, разрушении или функции нарушении тромбоцитов
• Лекарства (аспирин, метилдопа)
• Идиопатическая тромбоцитопеническая пурпура
• Тромбическая thrombocytopenic пурпура
• Диссеминированное внутрисосудистое свертывание
• Инфекция (особенно Neisseria, Rickettsia, Staphylococcus)
• Секвестрация селезёнки
• Лучевая терапия
• Миелофиброз
• Миелопролиферативные нарушения Сосудистая (обычно пальпируемая) Воспалительные сосудистые причины:
• Кожный васкулит
• Простой кожный васкулит ( вторичный лекарственный )
• пурпура Шейнляйн – Геноха
• Узловой периартериит
• Гранулематозный васкулит (гранулематоз Вегенера, гранулематозный аллергический ангиит)
• Кожный васкулит ассоциированный с коллагенозами
• Сосудистые заболевания (системная красная воланка, ревматический артрит)
• Гигантоклеточный артериит
• Смешанная криоглобулинемия
• Гиперглобулинемическая пурпура Невоспалительные сосудистые причины
• Травма
• Подострый бактериальный эндокардит
• Другие эмболические болезни
• Амилоидоз
• Экстраваскулярные и другие (может быть пальпируемой или не пальпируемой)
Кортикостероиды
• Токсины и яды
• Старческая пурпура
• Цинга
• проба Вальсальвы
• Псевдопурпура (Сладкий синдром, вишневые ангиомы, ангиокератомы, саркома Капоши ) Таблица 2: Клинические признаки некротического васкулита в зависимости от размера вовлеченных сосудов Мелкие сосуды Признаки • Крапивница
васкулит при гиперчувствительности
Пальпируемая Пурпура Болезни
пурпура Шейнляйн – Геноха
• Смешанная криоглобулинемия
• Васкулит, связанный с заболеваниями соединительной ткани
• Васкулит, связанный со злокачественными обазованиями
• Сывороточная болезнь и подобные реакции
•
Узлы, пузыри, или язвы Хроническая крапивница (уртикарный васкулит)
• Уртикарная продрома острого гепатита B Крупные сосуды Признаки
• Подкожные узлы, изъязвления, и экхимозы Болезни Узловой периартериит
• гранулематозный аллергический ангиит
• гранулематоз Вегенера •
Гигантоклеточный артериит
|
| |
| |
| Алексей |
Дата: Вторник, 10.03.2009, 08:25 | Сообщение # 3 |
  Украина Группа: Пользователи
Сообщений: 60
Статус: Offline |
КЛАССНЫЙ МАТЕРИАЛ |
| |
| |
| |
 Главная
Главная